
Contents
この記事を読んでできること
この記事では、CursorというAIエディタを用いてRを動かし傾向スコアマッチングと2値アウトカムのリスク差やオッズ比の推定を行う方法をご紹介します。
具体的には、一切コードを書かずに論文用のTableを作成し、さらにはマッチング前後の共変量バランスの可視化までAIへの指示のみで行います。
AIエディタを使えば、AIに指示を出すだけで全ての解析が完了し、綺麗なTableやFigureが完成します。
AIエディタについてもう少し詳しく知りたい方はこちらをご覧ください。
本記事と同じ解析環境を準備する手順は下記の記事で詳細に解説しています。
Cursorで傾向スコアマッチング
本記事の解析環境
- AIエディタ: Cursor (Version 0.45.11)
- AIモデル: claude-3.5-sonnet
以下のステップで分析を進めていきます。
- データセットの準備
- 傾向スコアの推定
- 傾向スコアの分布の確認(マッチング前)
- 傾向スコアマッチング
- 傾向スコアの分布の確認(マッチング後)
- 共変量のバランシングを確認
- 患者背景表の作成
- マッチングコホートを用いたオッズ比の推定
- マッチングコホートを用いたリスク差の推定
解析時にエラーが出て困ったときに読む記事もあるのでご安心ください。
それでは、早速分析を始めましょう。
データセットの準備
library(MASS)
df <- birthwt
| 変数名 | 説明 |
low |
出生時体重が2.5kg未満かどうかを示す指標(0 = 2.5kg以上、1 = 2.5kg未満) |
age |
母親の年齢(年) |
lwt |
最終月経時の母親の体重(ポンド) |
race |
母親の人種(1 = 白人、2 = 黒人、3 = その他) |
smoke |
妊娠中の喫煙状況(0 = 吸わない、1 = 吸う) |
ptl |
以前の早産の回数 |
ht |
高血圧の既往歴(0 = なし、1 = あり) |
ui |
子宮過敏性の有無(0 = なし、1 = あり) |
ftv |
妊娠初期の医師の診察回数 |
bwt |
出生時体重(グラム) |
- アウトカム変数:low(出生時体重が2.5kg未満かどうかを示す)
- 曝露変数:smoke(妊娠中の喫煙状況を示す)
- 交絡因子:age, lwt, race, ptl, ht, ui, ftv
データの下準備として、ID列を作成します。このID列は、各患者を一意に識別するために使用されます。
df <- df %>%
mutate(patient_id = row_number()) %>%
select(patient_id, everything())
傾向スコアの推定
続いて、喫煙の有無を曝露因子として傾向スコアを推定します。
傾向スコアとは、共変量で条件付けられた時に曝露を受ける確率です。
一般的に、傾向スコアの推定には曝露を目的変数としたロジスティック回帰モデルを使用します。
今回の場合では、喫煙の有無 (smoke) を目的変数、年齢 (age)、体重 (lwt)、人種 (race)、早産歴 (ptl)、高血圧 (ht)、子宮刺激 (ui)、妊婦健診回数 (ftv)を説明変数としたロジスティック回帰モデルから得られた予測値が傾向スコアになります。
まずはロジスティック回帰モデルを構築します。
model <- glm(smoke ~ age + lwt + race + ptl + ht + ui + ftv, data=df, family=binomial())
summary(model)
続いて、データセットの各患者についてロジスティック回帰モデルを当てはめたときの予測値(傾向スコア)を推定しデータフレームに追加します。
df$propensity_score <- predict(model, type="response" )
propensity_score追加後のdfのイメージ
| ID | 年齢 | 体重 | 高血圧 | propensity_score |
|---|---|---|---|---|
| 1 | 65 | 75.2 | 1 | 0.824 |
| 2 | 42 | 62.5 | 0 | 0.312 |
| 3 | 58 | 80.1 | 1 | 0.756 |
| 4 | 35 | 55.8 | 0 | 0.145 |
| 5 | 70 | 68.4 | 1 | 0.678 |
傾向スコアの分布の確認(マッチング前)
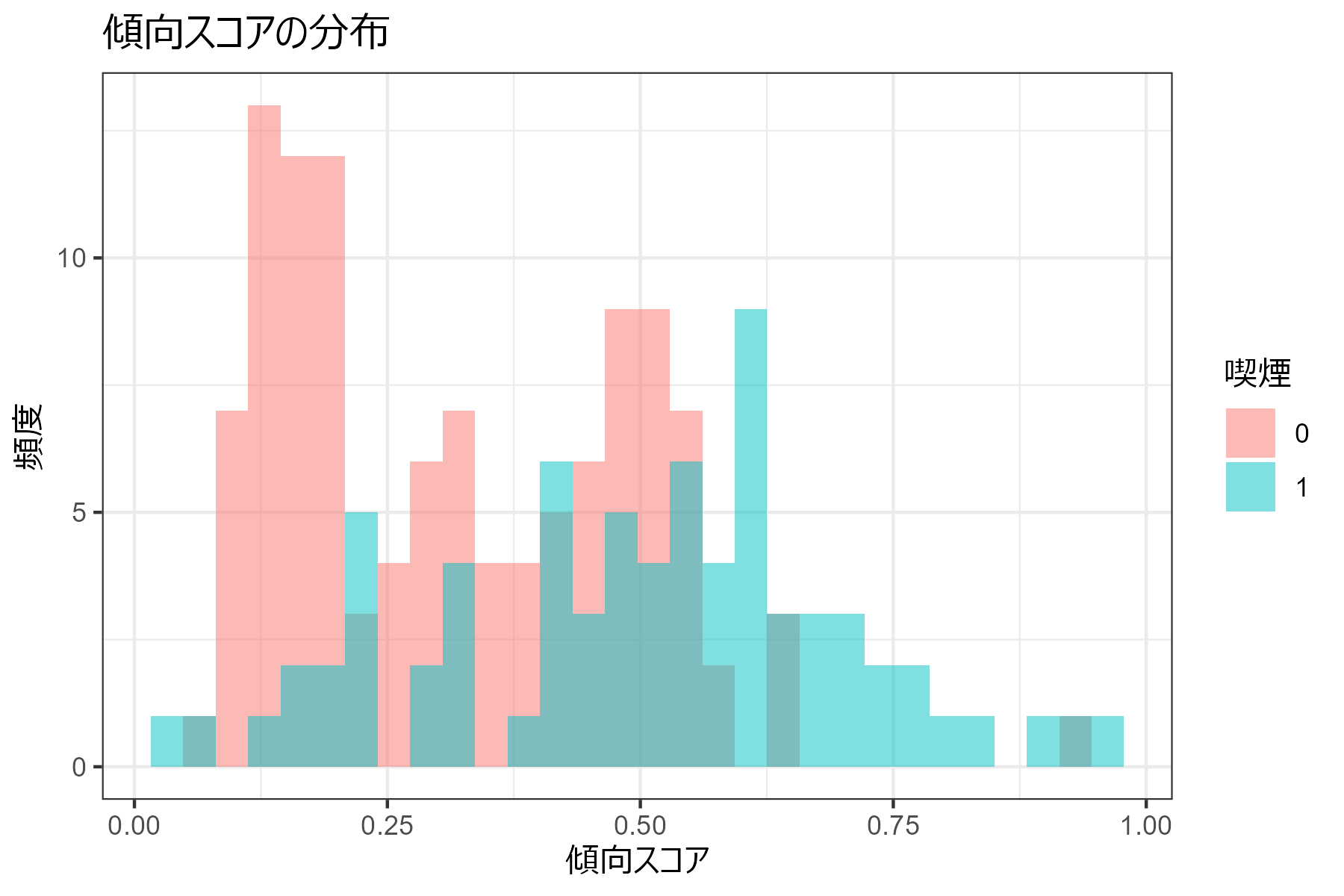
マッチング前の曝露群(喫煙あり)と対照群(喫煙なし)における傾向スコアの分布を確認してみます。
library(ggplot2)
ggplot(df, aes(x = propensity_score, fill = factor(smoke))) +
geom_histogram(position = "identity", alpha = 0.5) +
labs(title = "傾向スコアの分布",
x = "傾向スコア",
y = "頻度",
fill = "喫煙") +
theme_bw()
作成したプロットがこちらです。
傾向スコアマッチング
いよいよ、傾向スコアマッチングを行います。RではMatchItパッケージのmatchit関数がよく用いられます。
関数の中には、下記のようにマッチング方法や距離の計算方法などについて指定が必要なパラメータがいくつかあります。
matchit関数の主要パラメータ
| パラメータ | デフォルト値 | 主な選択肢 | 説明 |
| method | nearest |
|
マッチングのアルゴリズムを指定 |
| distance | glm |
|
マッチングの基準となる距離(類似度)の計算方法を指定 |
| link | logit |
|
距離尺度の推定に使用されるリンク関数を指定。"linear.{link}"とすると線形リンク関数が使用できる |
| caliper | NULL(使用しない) |
|
マッチングを許容する最大距離を指定 |
| std.caliper | TRUE |
|
キャリパーを標準偏差の単位で指定するかどうかを決定 |
| ratio | 1 |
|
処置群1名に対して対照群を何名マッチングさせるかを指定 |
| replace | FALSE |
|
1度マッチング相手となった症例が再度マッチング候補になることを許容する場合はTRUE |
下記のパラメータを指定してください。
method = nearest,
distance = glm,
link = linear.logit,
caliper = 0.2,
std.caliper = TRUE,
ratio = 1
library(MatchIt)
matchit_model <- matchit(smoke ~ age + lwt + race + ptl + ht + ui + ftv,
data = df,
method = 'nearest',
distance = 'glm',
link = 'linear.logit',
caliper = 0.2,
std.caliper = TRUE,
ratio = 1)
matched_data <- match.data(matchit_model)
下記2つはデフォルトの設定から変えています。
- link = 'linear.logit'(デフォルトは'logit')
- caliper = 0.2(デフォルトはNULL)
デフォルトのlink = `logit`の場合、傾向スコアそのものが距離の指標になります。この場合、傾向スコアがとり得る範囲 (0-1) のうち、真ん中 (0.5) 付近の0.1の差と、極端な値 (0や1) のときの0.1の差を同様に扱っています。
しかし、曝露を受ける確率が50%の症例と60%の症例と比べて、限りなく0に近い症例と10%の症例は性質が大きく異なると考えるのが自然です。
そのため、link = `linear.logit`とすることで距離の基準が0-1の範囲に強制されるのを防いでいます。
キャリパーは慣習的にlogit (傾向スコア) の標準偏差の0.2倍程度に設定されることが多いです。
キャリパーを小さくすると残余交絡を減らすことができますが、マッチングできる症例が減ってしまいマッチ後コホートの症例数が小さくなってしまいます。
キャリパーを大きくすると、マッチ後コホートの症例数が多くなりますが、残余交絡が大きくなりbiasedな結果に繋がります。
その他のパラメータはデフォルトの設定です。
今回のマッチングのアルゴリズムをまとめると
今回のように非復元の最近傍マッチングは多く用いられていますが、マッチング方法によるバイアスには様々議論があり、必ずしも最適とは限りません。
マッチングの方法の理論的側面を含めた実装方法については、高橋将宜著『統計的因果推論の理論と実装 ― 潜在的結果変数と欠測データ』(共立出版、2022年)が参考になりますので、ぜひご参照ください。
マッチング後のデータを確認してみましょう。
head(matched_data)
patient_id low age lwt race smoke ptl ht ui ftv bwt propensity_score distance weights subclass
85 1 0 19 182 2 0 0 0 1 0 2523 0.3310658 -0.70336837 1 17
87 3 0 20 105 1 1 0 0 0 1 2557 0.5827781 0.33418841 1 30
89 5 0 18 107 1 1 0 0 1 0 2600 0.6771450 0.74068213 1 39
92 7 0 22 118 1 0 0 0 0 1 2637 0.5420976 0.16878987 1 5
94 9 0 29 123 1 1 0 0 0 1 2663 0.4509575 -0.19680277 1 52
95 10 0 26 113 1 1 0 0 0 0 2665 0.5183271 0.07334119 1 1
| 変数名 | 説明 | 備考 |
| smoke | 処置変数(1/0) | 喫煙の有無 |
| age | 年齢 | マッチングに使用した共変量 |
| lwt | 体重 | マッチングに使用した共変量 |
| race | 人種 | マッチングに使用した共変量 |
| ptl | 既往歴 | マッチングに使用した共変量 |
| ht | 高血圧の有無 | マッチングに使用した共変量 |
| ui | 子宮刺激の有無 | マッチングに使用した共変量 |
| ftv | 妊婦健診回数 | マッチングに使用した共変量 |
| subclass | マッチングペアID | 新規追加列:マッチングされたペアを識別する番号 |
| distance | プロペンシティスコア | 新規追加列:ロジスティック回帰で算出された予測確率 |
| weights | マッチングの重み | 新規追加列:1:1マッチングの場合はすべて1 |
傾向スコアの分布の確認(マッチング後)
ggplot(matched_data, aes(x = propensity_score, fill = factor(smoke))) +
geom_histogram(position = "identity", alpha = 0.5) +
labs(title = "マッチング後の傾向スコアの分布",
x = "傾向スコア",
y = "頻度",
fill = "喫煙") +
theme_bw()
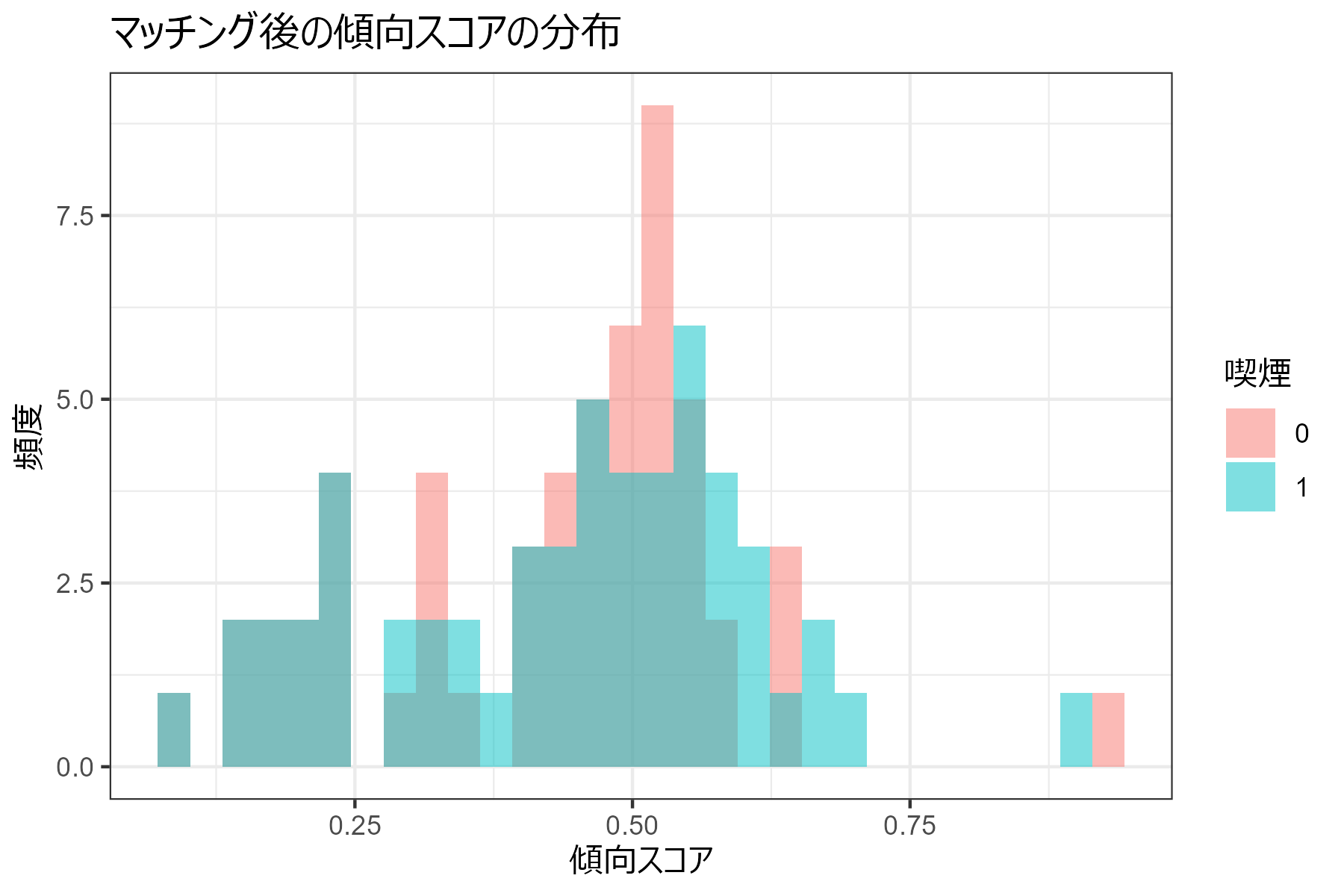
ペアが作れなかった患者が除外されたことで、マッチング後の傾向スコアの分布はマッチング前の分布と比較して、より均一になっていることがわかります。
共変量のバランシングの確認
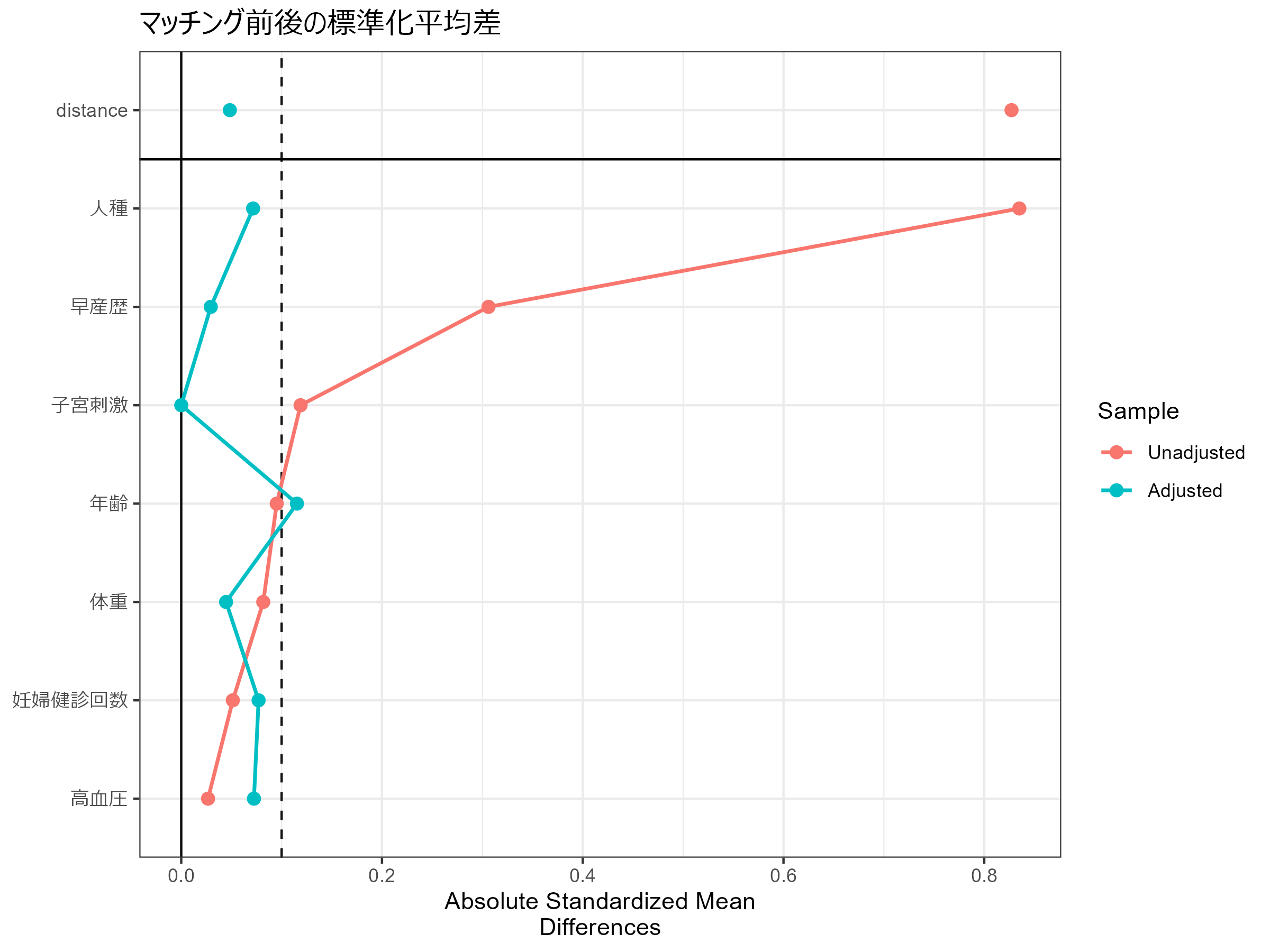
次に、cobaltパッケージのlove.plot関数を用いて、マッチング前後のコホートについて、age, lwt, race, ptl, ht, ui, ftvの標準化平均差を可視化してみます。
library(cobalt)
love.plot(matchit_model,
stats = "mean.diffs",
thresholds = c(m = 0.1),
var.order = "unadjusted",
binary = "std",
abs = TRUE,
var.names = c(age = "年齢",
lwt = "体重",
race = "人種",
ptl = "早産歴",
ht = "高血圧",
ui = "子宮刺激",
ftv = "妊婦健診回数"),
line = TRUE,
title = "マッチング前後の標準化平均差") +
theme_bw() +
theme(text = element_text(family = "HiraKakuProN-W3"))
赤色がマッチング前、青色がマッチング後のコホートにおける共変量の標準化平均差です。
この値は0に近いことが理想で、0.1を超えないことが望ましいとされています。
マッチング後は標準化平均差が小さくなっており、ほぼ0.1以内におさまっています。
患者背景表の作成
続いて、マッチング前・後それぞれの患者背景表を作成してみます。
群分け変数: smoke
連続変数: age, lwt
カテゴリカル変数: race, ptl, ht, ui, ftv
library(tableone)
vars <- c("age", "lwt", "race", "ptl", "ht", "ui", "ftv")
cat_vars <- c("race", "ptl", "ht", "ui", "ftv")
tab1 <- CreateTableOne(vars = vars,
strata = "smoke",
data = df,
factorVars = cat_vars)
print(tab1, showAllLevels = TRUE)
# マッチング後の患者背景表
tab2 <- CreateTableOne(vars = vars,
strata = "smoke",
data = matched_data,
factorVars = cat_vars)
print(tab2, showAllLevels = TRUE)
見やすくなるようにkable関数で表を整えて出力します。
マッチング前の患者背景表
| |0 |1 |p |test |
|:---------------|:--------------|:--------------|:------|:----|
|n |115 |74 | | |
|age (mean (SD)) |23.43 (5.47) |22.95 (5.05) |0.545 | |
|lwt (mean (SD)) |130.90 (28.43) |128.14 (33.79) |0.546 | |
|race (%) | | |<0.001 | |
|1 |44 (38.3) |52 (70.3) | | |
|2 |16 (13.9) |10 (13.5) | | |
|3 |55 (47.8) |12 (16.2) | | |
|ptl (%) | | |0.066 | |
|0 |103 (89.6) |56 (75.7) | | |
|1 |10 ( 8.7) |14 (18.9) | | |
|2 |2 ( 1.7) |3 ( 4.1) | | |
|3 |0 ( 0.0) |1 ( 1.4) | | |
|ht = 1 (%) |7 ( 6.1) |5 ( 6.8) |1.000 | |
|ui = 1 (%) |15 (13.0) |13 (17.6) |0.519 | |
|ftv (%) | | |0.155 | |
|0 |55 (47.8) |45 (60.8) | | |
|1 |35 (30.4) |12 (16.2) | | |
|2 |19 (16.5) |11 (14.9) | | |
|3 |3 ( 2.6) |4 ( 5.4) | | |
|4 |3 ( 2.6) |1 ( 1.4) | | |
|6 |0 ( 0.0) |1 ( 1.4) | | |
マッチング後の患者背景表
| |0 |1 |p |test |
|:---------------|:--------------|:--------------|:-----|:----|
|n |55 |55 | | |
|age (mean (SD)) |22.85 (4.94) |23.44 (5.30) |0.553 | |
|lwt (mean (SD)) |131.36 (30.37) |129.85 (32.13) |0.801 | |
|race (%) | | |0.887 | |
|1 |34 (61.8) |35 (63.6) | | |
|2 |8 (14.5) |9 (16.4) | | |
|3 |13 (23.6) |11 (20.0) | | |
|ptl (%) | | |0.951 | |
|0 |48 (87.3) |47 (85.5) | | |
|1 |5 ( 9.1) |6 (10.9) | | |
|2 |2 ( 3.6) |2 ( 3.6) | | |
|ht = 1 (%) |4 ( 7.3) |3 ( 5.5) |1.000 | |
|ui = 1 (%) |6 (10.9) |6 (10.9) |1.000 | |
|ftv (%) | | |0.156 | |
|0 |23 (41.8) |32 (58.2) | | |
|1 |20 (36.4) |9 (16.4) | | |
|2 |10 (18.2) |10 (18.2) | | |
|3 |1 ( 1.8) |3 ( 5.5) | | |
|4 |1 ( 1.8) |1 ( 1.8) | | |
ここまでで、傾向スコアマッチング、傾向スコアの分布の確認、共変量のバランシングの確認、患者背景表の作成が完了しました。
いよいよ、マッチング後のコホートを用いて解析を行います。
オッズ比の推定
傾向スコアマッチング後の解析ではロバスト分散推定を行うことが推奨されています。
2値アウトカムの解析においてロバスト分散推定を行うためには、geepackパッケージのgeeglm関数が便利です。
オッズ比を推定したいときはlink="logit"を指定します。
library(geepack)
gee_model <- geeglm(low ~ smoke,
data = matched_data,
id = subclass,
family = binomial(link = "logit"),
corstr = "independence")
geeglmの結果のサマリ作成には、broomパッケージのtidy関数が便利です。
library(broom)
model_summary <- tidy(gee_model, conf.int = TRUE, exp = TRUE) %>%
select(term, estimate, conf.low, conf.high, p.value) %>%
rename(odds_ratio = estimate)
print(model_summary)
# A tibble: 2 × 5
term odds_ratio conf.low conf.high p.value
1 (Intercept) 0.279 0.147 0.529 0.0000926
2 smoke 1.89 0.813 4.40 0.139
傾向スコアマッチングにより交絡因子の調整は行われているので、解析モデルは説明変数が曝露因子(喫煙の有無)のみの単変量解析を行っています。
マッチング後も共変量のバランシングが不十分な因子がある場合は、さらに上記のモデルに共変量を加えて多変量解析にすることで、残余交絡によるバイアスを取り除くことが期待されます。
ただし、マッチングに用いた因子は全て多変量回帰モデルに含めるようにしてください。マッチングに用いた因子以外をモデルに含めたり、一部のみを使用すると、他の因子のバランスを損なう可能性があることが報告されています [1]。
[1] Shinozaki T, Nojima M. Misuse of Regression Adjustment for Additional Confounders Following Insufficient Propensity Score Balancing. Epidemiology. 2019;30(4):541-548.
リスク差の推定
geeglm関数でリスク差を推定したいときはlink="identity"を指定します。
gee_model_rd <- geeglm(low ~ smoke,
data = matched_data,
id = subclass,
family = gaussian(link = "identity"),
corstr = "independence")
model_summary_rd <- tidy(gee_model_rd, conf.int = TRUE) %>%
select(term, estimate, conf.low, conf.high, p.value) %>%
rename(risk_difference = estimate)
print(model_summary_rd)
# A tibble: 2 × 5
term risk_difference conf.low conf.high p.value
1 (Intercept) 0.218 0.109 0.327 0.0000894
2 smoke 0.127 -0.0386 0.293 0.133
これで、傾向スコアマッチング、共変量バランシングの確認、患者背景表の作成、オッズ比・リスク差を推定することができました。

